
Engaging a Notified Body under the IVDR: What legacy manufacturers need to know about the pre-application process
The IVDR transition includes structured processes IVD manufacturers must follow to engage with Notified Bodies. Many IVD manufacturers are interacting with Notified Bodies for the first time and must understand what is required to ensure a successful partnership. In this article, Dr. Oliver Eikenberg clarifies what manufacturers must prepare and when to engage a Notified Body for IVDR CE Marking.
To obtain CE Marking under (EU) 2017/746 (IVDR), legal manufacturers of IVDs classified as Class A sterile, Class B, C, or D must sign a formal application with an accredited Notified Body (NB). Anyone familiar with this process understands the complexity of selecting a NB and following the stages of the NB application, which can vary between NBs. With deadlines approaching to engage a NB in line with IVDR transition requirements, IVDR companies must understand the NB pre-application process to meet their final IVDR deadline. This is especially important for IVD manufacturers marketing their IVDs as legacy devices who are engaging independent NB reviewers for the first time.
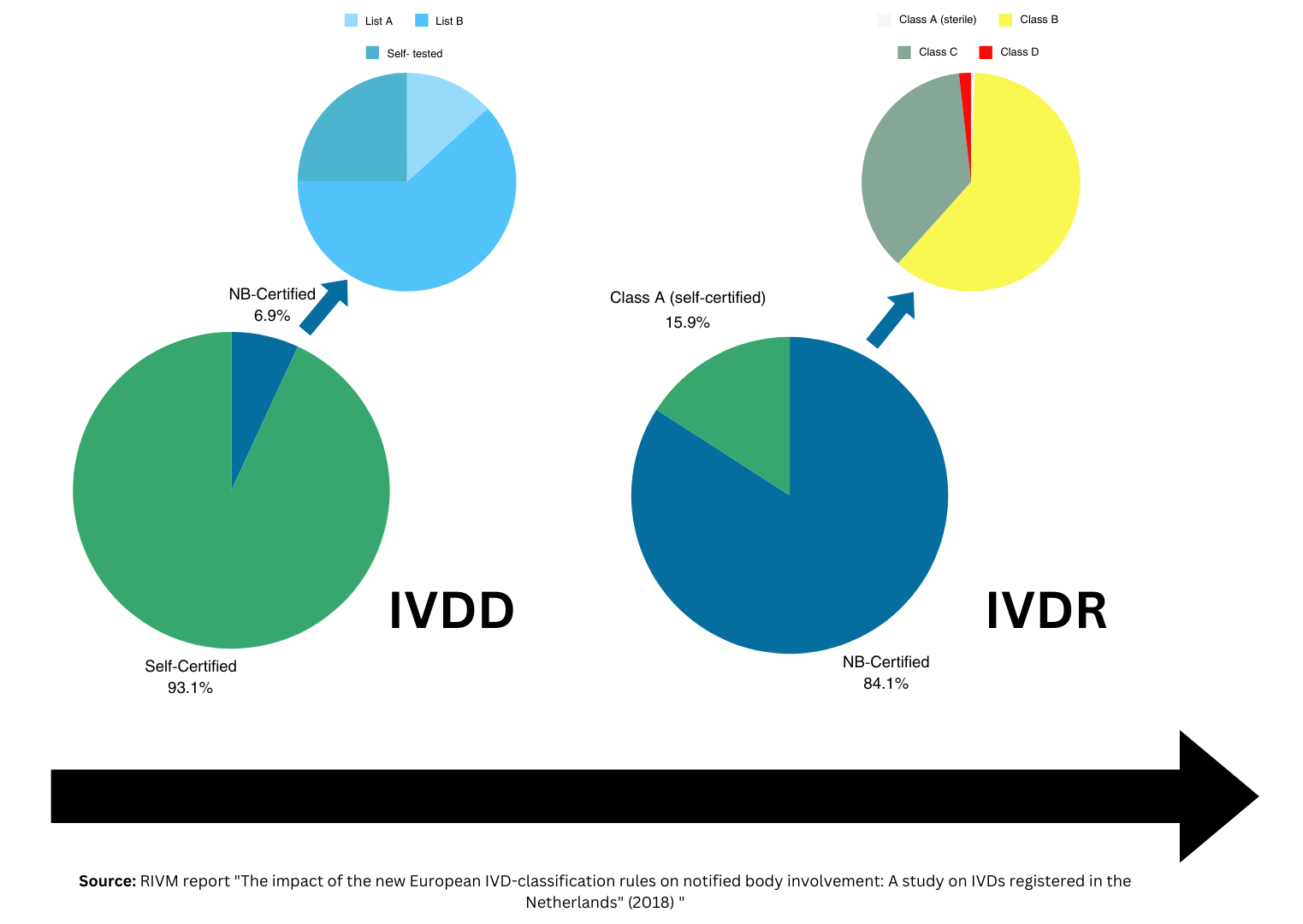
Legacy device manufacturers should be aware of the latest amendment to the IVDR, regulation EU 2024/1860, which introduces a series of important updates. These include the phased introduction of EUDAMED, new supply chain obligations, and revised transitional provisions for legacy IVD devices as follows:
Understanding the limitations, deadlines, and specific requirements of this amendment is essential for all IVD manufacturers who wish to continue operating within the EU market. For legacy IVD devices, it includes new deadlines and different pre-application steps, which should be carefully considered.
Notified Body pre-application process for IVD manufacturers
Team-NB members published a consensus document on MDR Certification, which describes in detail the pre-application, application, and post-application processes for medical device manufacturers. This consensus document applies to legacy medical devices (pursuant to Article 120) transitioning to MDR, as well as medical devices that are new to the market and have not previously been certified under the Directives. Though this is not a binding document nor yet published for IVDs, it supplies a detailed view of the general controls and obligations of NBs, which are expressed in “Appendix A: List of data/documents to be submitted by the manufacturer at various phases of the process.” The overall approach of the application stages will likely be the same for IVDs.
Your first interactions with a NB include lodging an application and signing an agreement with the NB, usually four months later. These steps must be completed several months before the NB requests complete data to perform their Technical Documentation Assessment (TDA) review. The preliminary stages enable the NB to balance internal resources and deliver timely service (review) before the final transition deadline ends.
What does an IVD manufacturer need to lodge an application with a NB?
Detailed obligations or details about pre-stage TDA review processes do not exist because these stages were not considered when the MDR or IVDR were drafted. However, the EU Commission has published a Q&A on practical aspects related to the implementation of the extended transitional period provided for in the IVDR, as amended by Regulation (EU) 2024/1860 for interpretation purposes.
This Q&A clarifies that a full review of the application (full TDA) is not required by the NB before lodging an application or signing the written agreement. This is reasonable, as the IVD manufacturer might need more time to refine their Technical Documentation. It also clarifies that the application must allow the NB to qualify the products as IVDs, identify their classification under the IVDR, and identify the chosen conformity assessment procedure. When lodging the application, the manufacturer should provide a timeline for the expected submission. All this information demonstrates that the IVD manufacturer understands and has implemented key elements of the IVDR before lodging the application with the NB.
Classification and intended purpose
Classification is a frequent problem in the pre-application request, as it can only be confirmed if the intended purpose is clearly expressed in the labeling in accordance with the criteria from IVDR Annex 20.4.1 (c). For objective evidence, the intended purpose should be reflected in the instructions for use (IFU) and labelling. Even if manufacturers can continue to sell their IVDs under IVDD, it is strongly recommended to prepare the new intended purpose and draft the proposed IFU in controlled documents. Otherwise, a classification cannot be confirmed, and the application will not be signed by the NB. For a deeper dive into this topic, read our blog on the role of intended purpose under the IVDR.
QMS outputs
According to Appendix A from the Team NB consensus document, information on the SRN number, UDI-DI, EMDN code, selected EU Authorized Representative (EU REP), and the involved “crucial suppliers and/or critical subcontractors” are also required during the pre-application stage. These are outputs from basic QMS requirements, which every IVD manufacturer is required to implement by 26th May 2025. However, if the QMS procedures are not actively understood and completed by the IVD manufacturers in time, the output of required information for the NB will not be adequate. Even if IVD manufacturers are audited months later by the NBs, missing evidence for an IVDR-ready QMS by 26th May 2025 could still be a significant non-conformity. Note that inside a QMS, it is easy for a NB to identify when and what has been implemented and effective.
Technical documentation
Notably, the Team NB consensus document mentions that the NB may request a summary/section of or the full Technical Documentation as part of the application. This is to gather enough information about the devices to verify the qualification as devices, their respective classification, and the chosen conformity assessment procedure. The document further states that “based on the application review, the NB will decide whether to accept the application or refuse the application (only after the signing of the contract).” It means that NBs might not be willing to take your application and then it would be up to you to apply to another NB. This could significantly change your time to market significantly, particularly if NB application deadlines have already lapsed.
IVDR adherence is essential in the pre-application stage
As a conclusion, many elements of the IVDR should be understood and implemented in an adequate way some time before the application with the NB is lodged and the agreement with the NB is signed. This is especially true for the intended purpose, classification, and basic QMS procedures addressed in IVDR Article 10 (including EUDAMED registration and labelling, UDI, PMS, PE process, and vigilance). Be advised that your NB may request documents that demonstrate how you followed the QMS procedures in accordance with IVDR requirements. Manufacturers should follow processes and basic requirements according to the most recently published state-of-the-art standards and interpretations, such as safety, labelling, and risk management. For example, new symbols or labelling information for your IVD identify adequate compliance to applicable safety regulations or classification in line with latest EMDN codes.
It is too early in the transition timeline to anticipate what detailed questions could arise during the NB process to lodge an application or sign an agreement. However, IVD manufacturers should be proactive and check their documents against IVDR requirements as early as possible to avoid potential non-conformities or rejection of their application.
Pure Global’s experienced IVDR experts can help you navigate Notified Body search and selection and the pre-application stage. We assist with IVDR classification confirmation, QMS process implementation, documentation and labeling review, and more. Contact us to learn how we can help.



Let's Talk,
Anywhere You Are.
Whether looking for more information or ready to partner with us, we're here to guide you through every step of the regulatory process.
Contact us