
What are the challenges under IVDR?
IVD manufacturers cite the new classification system, detailed definitions for intended purposes, and the need for verification of clinical evidence as key challenges under the IVDR.
What are the challenges under IVDR?
According to statements from the EU Commission, the overall structure and approach of the system have not changed between the IVDD and IVDR. However, IVD manufacturers have been complaining for years about the challenges the IVDR brings. These challenges mainly stem from the new classification system, detailed definitions for intended purposes, and the need for verification of clinical evidence.
How are these 3 elements linked to each other?
Under the IVDD, there was no risk-based classification system, and IVDs were classified based on a positive listing that reflected state-of-the-art technology and knowledge from 1998. Since this system became inadequate, the EU decided to align with the classification system recommended by the Global Harmonization Task Force (GHTF) in 2008, which is already used in many countries. The new GHTF-based system classifies IVDs into four risk-based classes, introducing risk classification and implementation rules to determine the level of regulatory conformity assessment required for an IVD.
As part of this, classification rules are now linked to the intended purpose and claims defined by the manufacturer. For example, a device intended for the diagnosis of cancer or blood grouping is classified in a higher risk class compared to IVDs where the diagnosis does not have a serious impact on patient health.
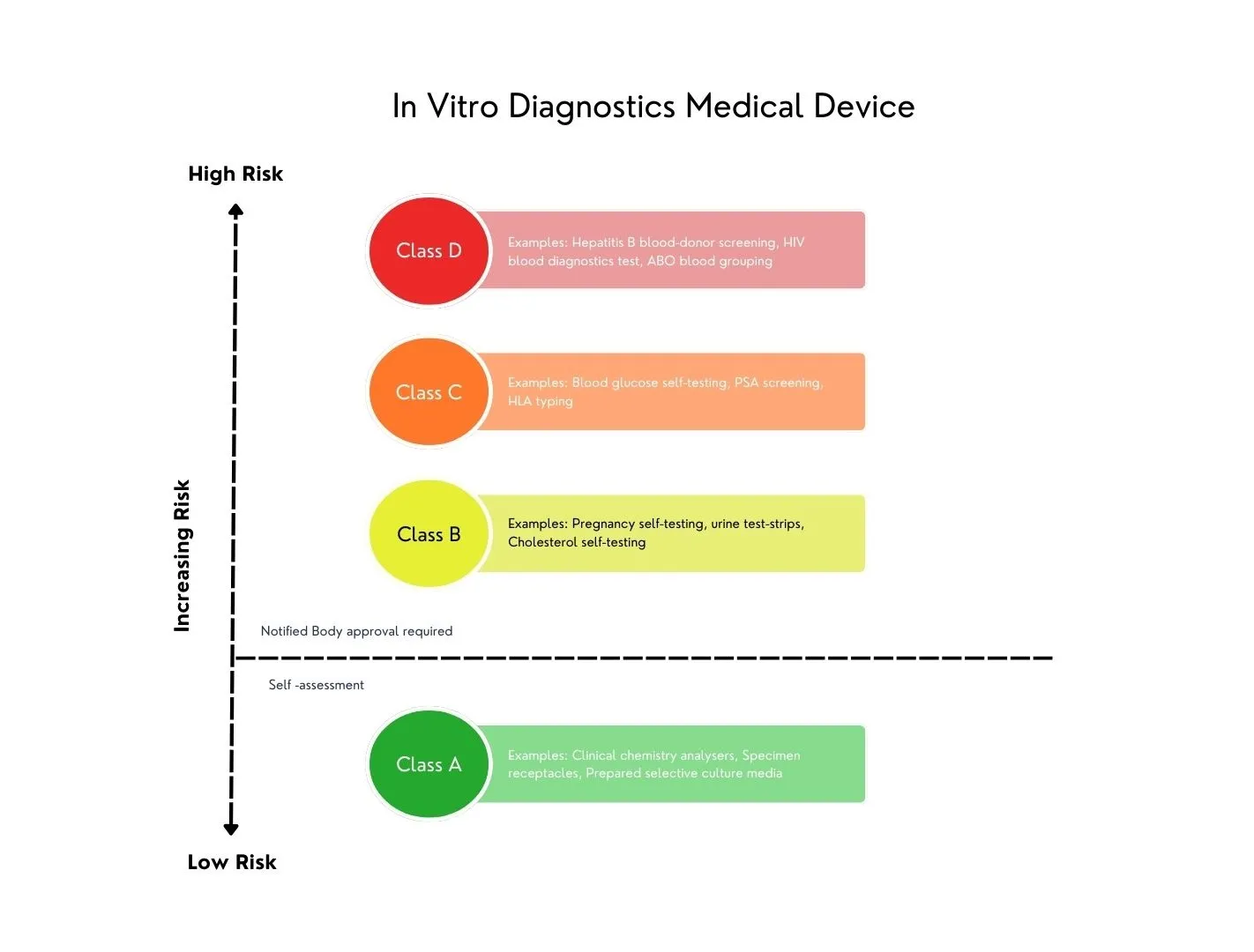
This shift means that the intended purpose must go beyond merely reflecting the analytical measurement. It must align with the claims and indications defined by the manufacturer. Furthermore, these claims must be verified as part of the clinical evidence, which includes scientific validity, analytical performance, and clinical performance, through a performance evaluation process (separate plan and report).
One of the main challenges in this process is the transition from a purely analytical intended purpose, such as 'intended to measure in EDTA plasma,' to a more comprehensive definition that includes the target group, indication, and use scenario. For example, 'intended to measure in EDTA plasma on the automated analyzer [device name] for human patients aged [age range], for cancer screening.' This requires a high level of detail and clarity.
Why is this impacting manufacturers?
IVDs now require clear verification and documentation of all claims in their intended purpose through performance data, which may limit sales if clinicians only use them for the specified purpose. Additionally, for combined devices, such as IVD kits used on automated analyzers, the combined use must be clearly stated and verified. If manufacturers already have sufficient data to support these claims, the transition from IVDD to IVDR may not be a significant challenge. However, lacking this data could lead to significant discussions between sales and regulatory departments about what should be included in the intended purpose claims. This will also require substantial work to verify the claims, aligning them with classification and clinical evidence, which may include clinical performance studies to confirm their use for the target group and intended use scenario.
In summary, the changes in the classification system for IVDs have a significant impact on the intended purpose and clinical evidence. For medical devices, a similar risk-based classification system has been part of the MDD, which is why medical device manufacturers are accustomed to it, whereas IVD manufacturers are not yet used to it. These challenges continue to affect many IVD manufacturers, and often they only become apparent when their data are first reviewed and assessed by an independent Notified Body. As with many other things, good planning and awareness are key to meeting regulatory requirements. With proper preparation, CE marking under IVDR is not as difficult as it may seem.
As IVD manufacturers navigate the complexities of IVDR compliance, expert guidance and the use of advanced AI tools can make a significant difference, saving both time and money. Reach out to Pure Global for assistance in overcoming regulatory challenges and ensuring your product meets all necessary IVDR requirements.
Latest Blog Content
Explore our collection of articles, success stories, and regulatory updates, designed to help you take your product global.




Let's Talk,
Anywhere You Are.
Whether looking for more information or ready to partner with us, we're here to guide you through every step of the regulatory process.
Contact us