
Conformity under the IVDR requires a rigorous approach to clinical evidence quality. In this article, Dr. Oliver Eikenberg explains the importance of performance evaluation processes and documentation to ensure adequate clinical evidence for EU IVDR compliance.
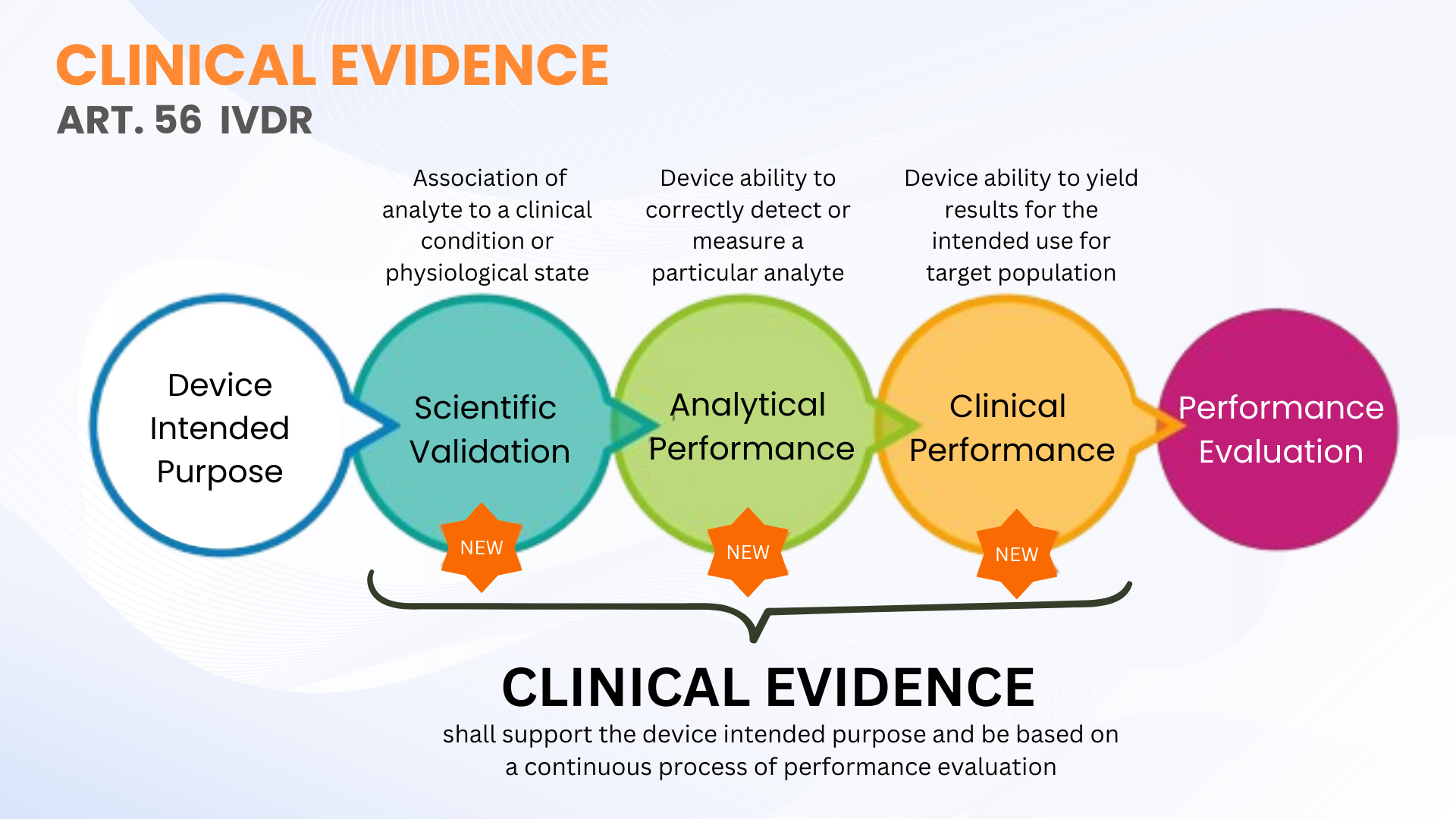
Clinical evidence is not a new requirement under In Vitro Diagnostic Devices Regulation (EU IVDR 2017/746); however, it is more clearly defined in contrast to the IVDD. Article 56 and Annex XIII of the IVDR define clinical evidence as a continuous process of performance evaluation (PE) for IVDs that includes scientific validity, analytical performance, and clinical performance. In simpler terms, clinical evidence comprises the data and findings from the performance evaluation to demonstrate that your IVD is safe and performs according to its intended purpose.
Considering the IVDR expanded criteria for intended purpose and introduced a more rigorous approach to safety in general, many IVDR manufacturers are wondering how far they need to go to ensure they present adequate clinical evidence, especially as IVDR transition deadlines approach.
However, many IVD manufacturers are still underestimating the need for a careful PE process as well as the benefits of providing clearly appraised and analyzed PE data and an adequate level of information to Notified Bodies (NBs). Because clinical evidence is the overall decision criteria for a safe and effective device, thorough PE documentation, process, and risk-based accurate data analysis are the keys to passing an NB review.
Performance evaluation (PE) is the mechanism that supports your clinical evidence. It should be a well-structured, transparent, iterative, and continuous process by which data are assessed and analyzed to demonstrate the scientific validity, analytical performance, and clinical performance of the device for its intended purpose. It must be part of the manufacturer’s QMS and occurs throughout the complete life cycle including post-market.
Like many other processes, PE starts with planning. The Performance Evaluation Plan (PEP) must define the data collection methods, timelines, acceptance criteria, and personnel responsibilities. PE data collection from PE testing is then conducted according to the PEP. Available scientific or PE data are collected and evaluated (appraised) based on the defined data analysis, the conclusions of which will inform the Performance Evaluation Report (PER). Post-Market activities cover the continuous monitoring and evaluation of the state-of-the-art (e.g., standards, Post Market Performance Follow-up (PMPF), activities documented in the manufacturer's PMPF Report and Periodic Safety Update Report (PSUR), as applicable). Risk Management is the core of all these activities and should be included in each step.
MDCG 2022-2 illustrates the PE process in a simplified graph:

source: MDCG 2022-2
As stated, clinical evidence for IVDs can be established through the collection of PE data from scientific validity, analytical performance, and clinical performance. The evidence should be collected and documented to a level that allows a qualified Technical Documentation assessment (TDA by Notified Body) of the device’s safety and whether it achieves the intended clinical benefit(s) when used as intended by the manufacturer. This does not necessarily mean that clinical performance studies must be conducted. Sometimes, scientific validity assessment through qualified evaluators and analytical PE testing with an adequate number of clinical samples are sufficient.
The IVDR does not define specific numbers of clinical samples to be tested for certain IVD devices (except for some high-risk IVD parameters, where these are defined in Common Specifications). This flexibility allows IVD manufacturers to plan and conduct the PE testing appropriate for their device. However, manufacturers should plan to scale their clinical performance study approach to support the clinical evidence according to the device’s risk classification. The higher the risk class, the more likely it is that clinical performance studies are needed to support the clinical evidence.
IVD manufacturers must define and submit a PEP in accordance with IVDR, Annex XIII, part A, section 1.1 before they conduct PE testing, as failure to do so could compromise the integrity of their test data. Without evidence that their PE testing followed the original PEP (and pre-defined acceptance criteria), NB reviewers may conclude that the PEP was revised after the fact to substantiate the test results. NB-auditors can easily verify and track when PEPs were released, how they were followed, and when PE testing was done. If this timeline is not clear, it usually raises questions, such as: Is the PE process not clearly defined? Why is it not documented as required under IVDR? Why are acceptance criteria and methods for PE data analysis not included? Therefore, to make your design and development, and CE Marking process more efficient, IVD companies should strictly follow the well-defined PE documentation processes and requirements in the IVDR.
Many IVD manufacturers struggle to implement a structured but agile and efficient PE process that does not limit innovation from Design & Development activities. A simple requirement to have a QMS procedure describing their PE activities or to have a clearly defined PE Plan before starting the PE activities is still a major challenge for many companies. This is often related to inadequate QMS processes, training, and understanding of PE requirements defined in the IVDR and interpretation guidelines, such as MDCG 2022-2.
Adequate documentation of PE results, conclusions, and rationales in the PER is critical to the success of manufacturer’s TDA as NB-auditors are not allowed to interpret any data on behalf of the manufacturer. If the documentation and rationales are not clearly expressed, this could result in a worst-case estimation by NB reviewers with the need to conduct additional clinical performance testing or PMPF studies. A good method to check the adequacy is to ask yourself if you would release your IVD device based on the PE data and objective evidence demonstrated in your PEP and PER. If you do not have the capabilities in house, qualified clinical evaluators can support the final writing of your PER to ensure it meets NB auditor requirements.
Manufacturers of legacy IVDs working with a revised and or expanded intended purpose under the IVDR might need to redefine their overall PE activities, scientific validity, analytical performance, and clinical performance testing to demonstrate the clinical evidence. In some cases, the PEP may need to be created retrospectively, if the PE process was not yet established at the time the device was designed and developed. If so, manufacturers should describe this retrospective approach clearly in their PE process and rewrite the PEP and PER in accordance with IVDR requirements and state-of-the art standards. Typically, substantial updates to the PEP and PER require detailed justifications and risk assessments for the conducted adaptations and changes. If this is not done, manufacturers may face significant questions from NB reviewers.
Many of these scenarios can be easily avoided if PE information is of superior quality and is clearly documented and verified. The implementation of collected Post-Market Surveillance (PMS) data can further support clinical evidence like a specific technology or marker. PMS data can also verify that the technology or device is well-established and that it has been well-controlled by the manufacturer. This further reduces the overall risk for the device, which contributes to the success of your NB review.
If you need support establishing PE processes, updating your QMS to IVDR, or help writing PE documentation, Pure Global can help. Our SMART software tools streamline the collection and appraisal of scientific data as well as selecting similar or equivalent devices on the EU market. Learn more about our performance evaluation support.
Explore our collection of articles, success stories, and regulatory updates, designed to help you take your product global.
Whether looking for more information or ready to partner with us, we're here to guide you through every step of the regulatory process.
Contact us


